






0
All
We use computational modeling to study emergent phenomena in condensed matter physics and to address challenges in the areas of energy and information technology.
The overarching theme of our research is to develop and apply predictive multiscale computational approach that combines formerly isolated methods to enable accurate and efficient estimations of materials properties at technologically relevant length/time scales.We comprehend the structure-property relationships of current state-of-the-art materials and then use these insights to guide the design of advanced materials. Research areas in our lab are in:
Explanation of Website Background Image: The background image on our website provides a vivid representation of our lab's three primary research areas. We have developed a deep neural network-based classical force field, facilitating large-scale molecular dynamics simulations of the PbTiO3/SrTiO3 superlattice. PbTiO3 is a prototypical ferroelectric material possessing spontaneous electric polarization. Within this superlattice, PbTiO3's unit cells exhibit electric dipoles, leading to the emergence of vortex structures - topological configurations in real space. To enhance the visual appeal of this snapshot, we have artistically blended it with elements from Van Gogh's iconic masterpiece, "Starry Night," using a specialized AI algorithm.




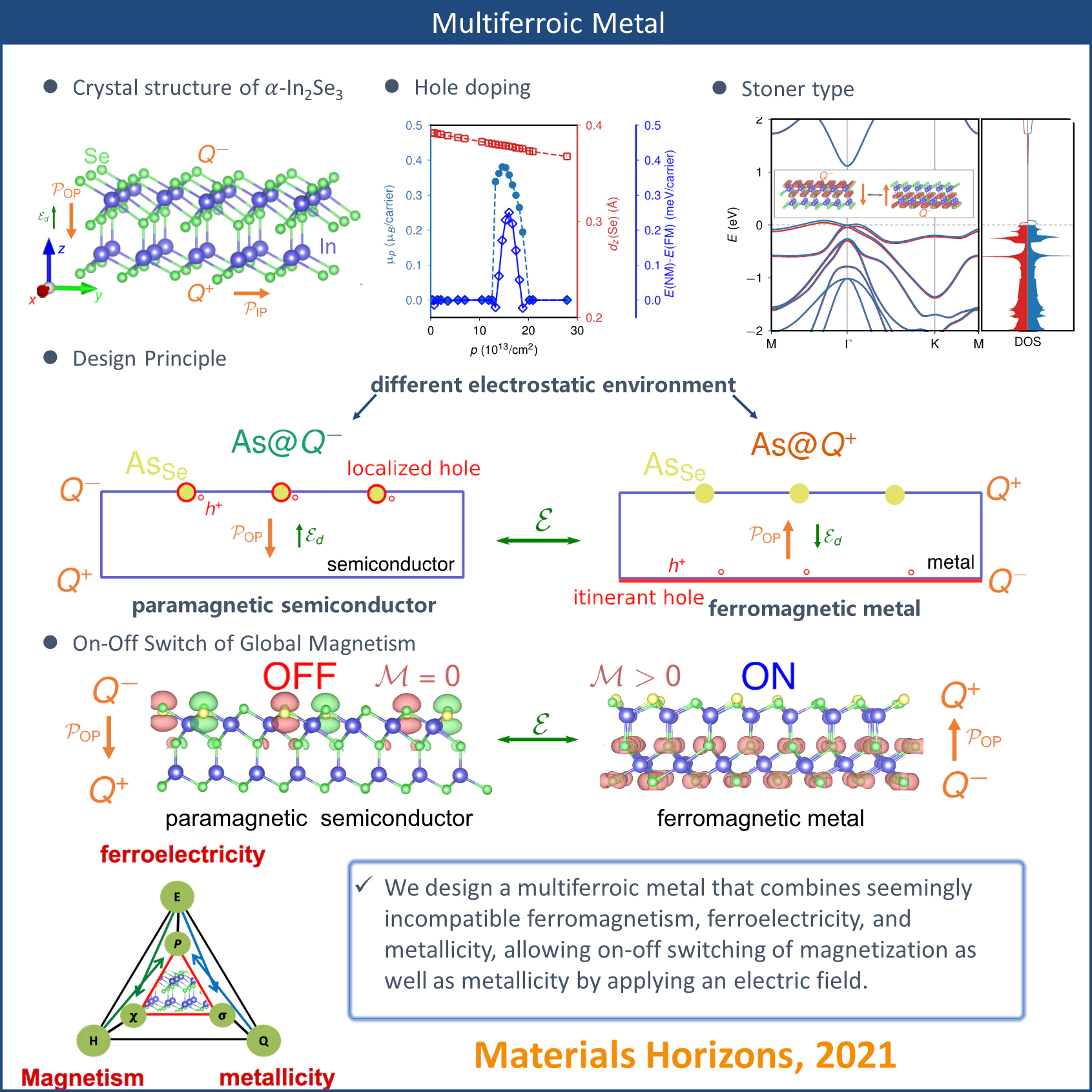
Taking advantage of the oppositely charged surfaces created by an out-of-plane polarization, the 2D magnetization and metallicity can be electrically switched on and off in an asymmetrically doped monolayer. The substitutional arsenic defect pair exhibits an intriguing electric field-tunable charge disproportionation process accompanied by an on–off switch of local magnetic moments. The charge ordering process can be controlled by tuning the relative strength of on-site Coulomb repulsion and defect dipole-polarization coupling via strain engineering. Our design principle relying on no transition metal broadens the materials design space for 2D multiferroic metals.
DOI: 10.1039/d1mh00939g
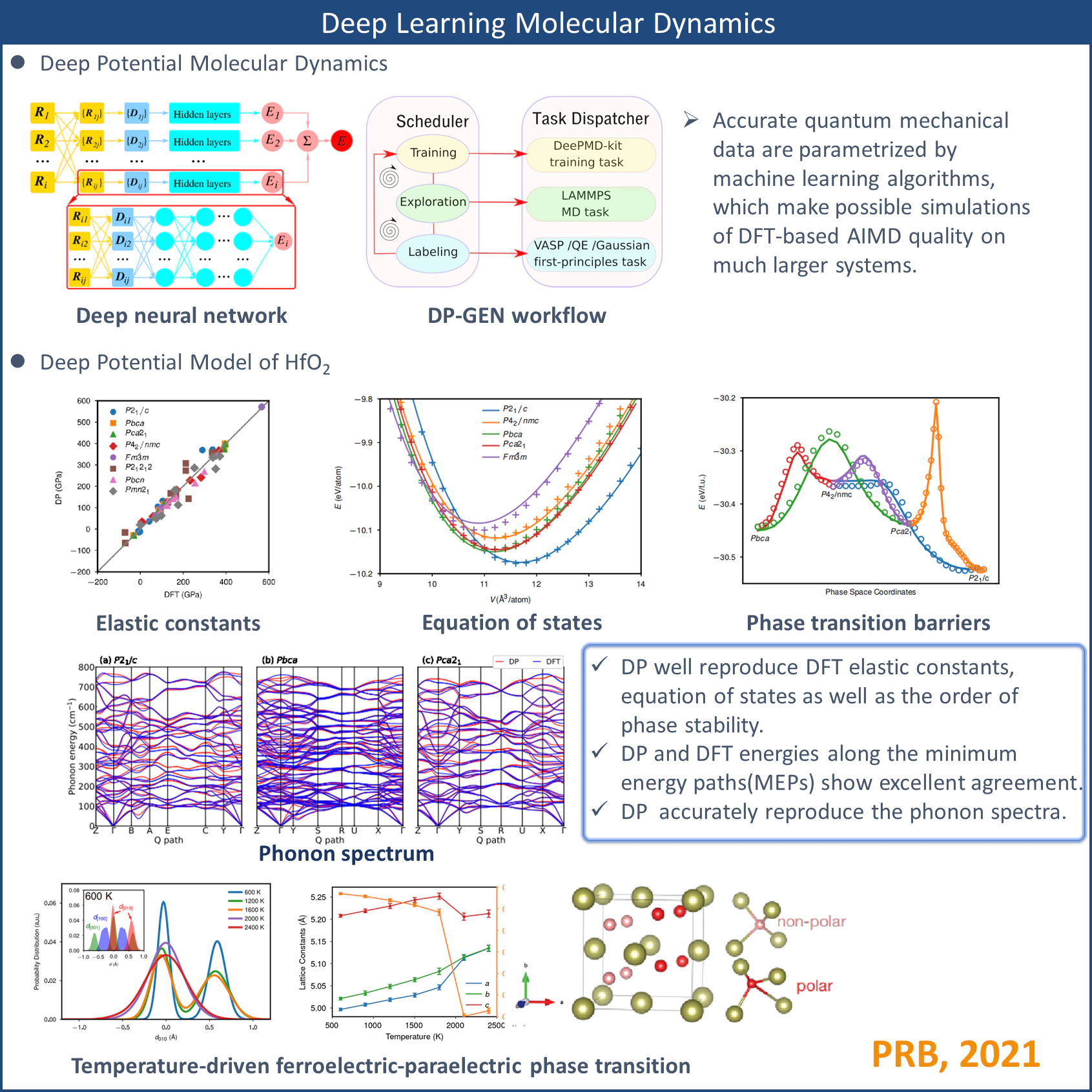
The model potential is able to predict structural properties such as elastic constants, equation of states, phonon dispersion relationships, and phase transition barriers of various hafnia polymorphs with accuracy comparable with density functional theory calculations. The validity of this model potential is further confirmed by the reproduction of experimental sequences of temperature-driven ferroelectric-paraelectric phase transitions of HfO2 with isobaric-isothermal ensemble molecular dynamics simulations. We suggest a general approach to extend the model potential of HfO2 to related material systems including dopants and defects.
DOI: 10.1103/PhysRevB.103.024108
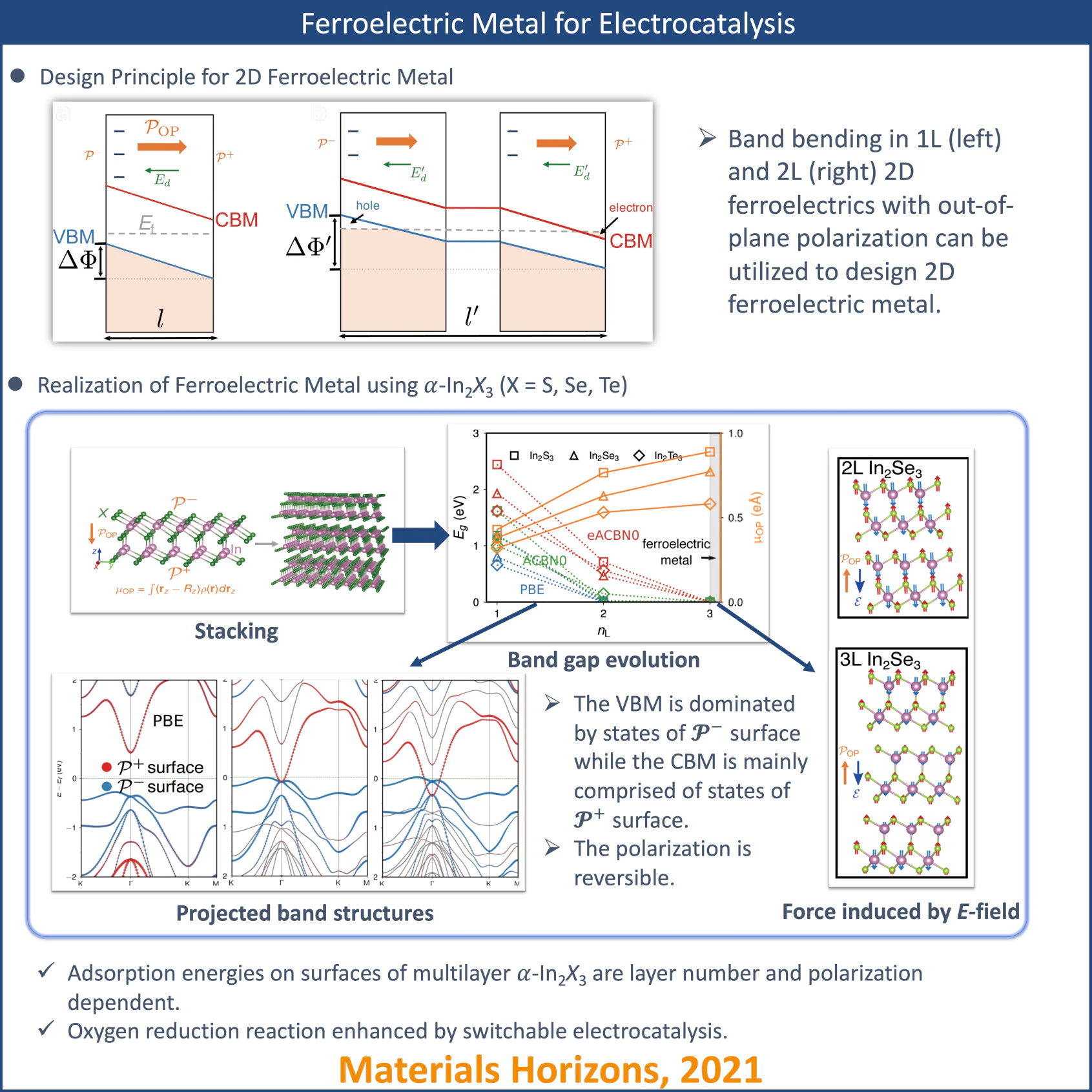
The coexistence of metallicity and ferroelectricity has been an intriguing and controversial phenomenon as these two material properties are considered incompatible in bulk. We clarify the concept of ferroelectric metal by revisiting the original definitions for ferroelectric and metal. Two-dimensional (2D) ferroelectrics with out-of-plane polarization can be engineered via layer stacking to a genuine ferroelectric metal characterized by switchable polarization and non-zero density of states at the Fermi level. We demonstrate that 2D ferroelectric metals can serve as electrically-tunable, high-quality electrocatalysts.
DOI: 10.1039/D1MH01556G

Long-range ferroelectric crystalline order usually fades away as the spatial dimension decreases, and hence there are few two-dimensional (2D) ferroelectrics and far fewer one-dimensional (1D) ferroelectrics. Due to the depolarization field, low-dimensional ferroelectrics rarely possess the polarization along the direction of reduced dimensionality. Here, using first-principles density functional theory, we explore the structural evolution of nanoribbons of varying widths constructed by cutting a 2D sheet of ferroelectric α-III2VI3 (III = Al, Ga, In; VI = S, Se, Te). We discover a one-dimensional ferroelectric nanothread (1DFENT) of ultrasmall diameter with both axial and radial polarization, potentially enabling ultra-dense data storage with a 1D domain of just three unit cells being the functional unit. The polarization in 1DFENT of Ga2Se3 exhibits an unusual piezoelectric response: a stretching stress along the axial direction will increase both the axial and radial polarization, referred to as the auxetic piezoelectric effect. Utilizing the intrinsically flat electronic bands, we demonstrate the coexistence of ferroelectricity and ferromagnetism in 1DFENT and a counterintuitive charge-doping-induced metal-to-insulator transition. The 1DFENT with both axial and radial polarization offers a counterexample to the Mermin–Wagner theorem in 1D and suggests a new platform for the design of ultrahigh-density memory and the exploration of exotic states of matter.
DOI: 10.1039/d3nh00154g

Salt-concentrated aqueous electrolytes show a wider electrochemical window than conventional aqueous electrolytes, yet still suffer from significant hydrogen evolution reaction (HER) at <1.9V versus Li†/Li. Introducing organic compounds was reported to alleviate HER, but all reported organic additives are flammable, inevitably compromising the safety property. Here, we report a new all-nonflammable-ingredient aqueous electrolyte via hybridizing with nonflammable methylurea. The structurally asymmetric methylurea molecules possessing both donor and acceptor functional groups regulate the hydrogen bonding network, resulting in peculiar nanoscale core-shell-like clusters. Such unique solution structure allows localized super-high salt concentration in the electrolyte and suppresses HER at 0.5V versus Li†/Li, achieving a 4.5V electrochemical window. Under a harsh testing condition with low electrolyte loading, no excess Li resource, no electrode precoating, and conventional aluminum current collectors, this electrolyte realizes a stable cycling of a rocking-chair NbO2|LiMn2O4 full cell (~175 Wh kg–1) without compromising the safety property.
DOI: 10.1016/j.joule.2022.01.002
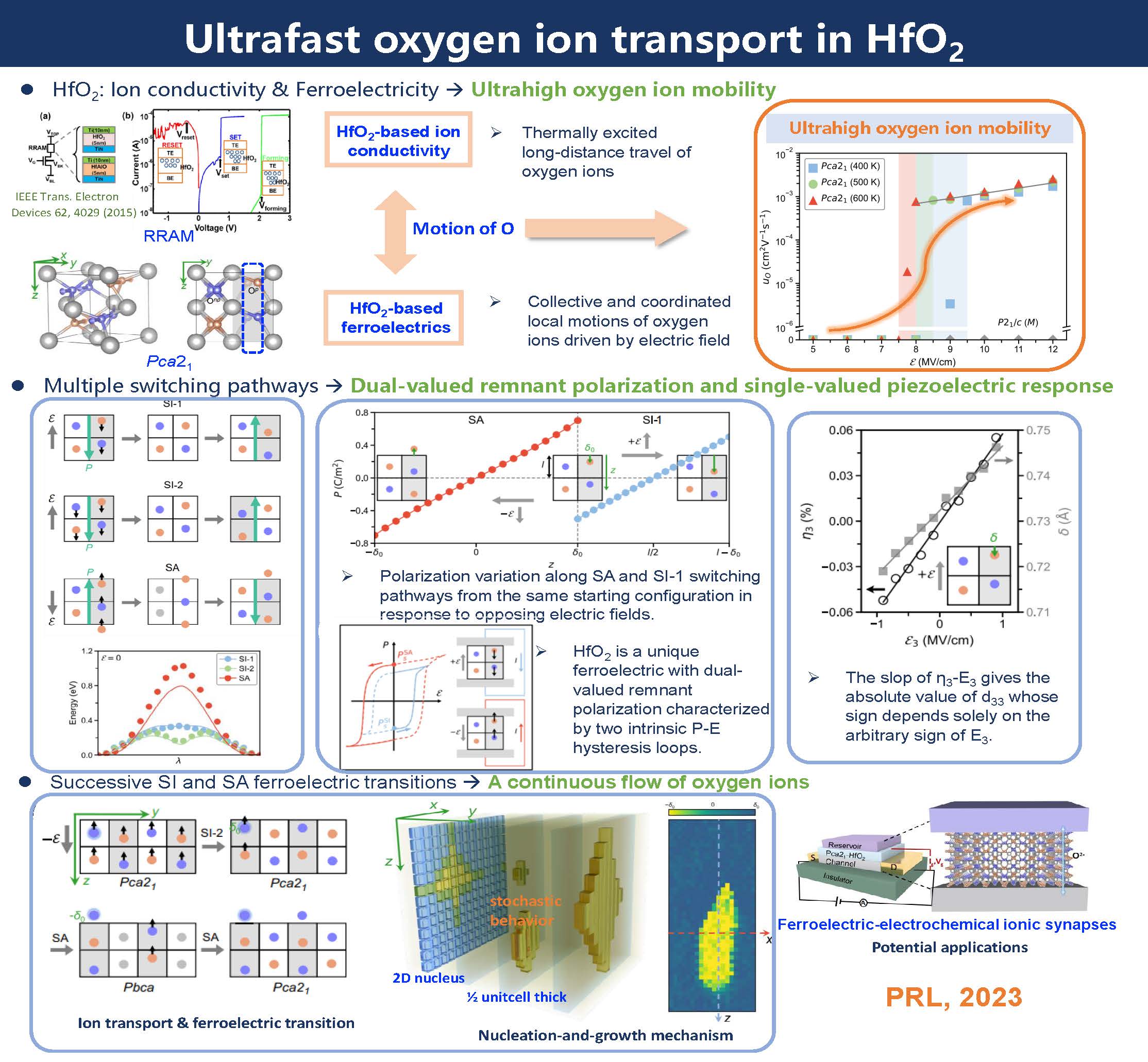
Ferroelectrics and ionic conductors are important functional materials, each supporting a plethora of applications in information and energy technology. The underlying physics governing their functional properties is ionic motion, and yet studies of ferroelectrics and ionic conductors are often considered separate fields. Based on first-principles calculations and deep-learning-assisted large-scale molecular dynamics simulations, we report ferroelectric-switching-promoted oxygen ion transport in HfO2, a wide band-gap insulator with both ferroelectricity and ionic conductivity. Applying a unidirectional bias can activate multiple switching pathways in ferroelectric HfO2, leading to polar-antipolar phase cycling that appears to contradict classical electrodynamics. This apparent conflict is resolved by the geometric quantum-phase nature of electric polarization that carries no definite direction. Our molecular dynamics simulations demonstrate bias-driven successive ferroelectric transitions facilitate ultrahigh oxygen ion mobility at moderate temperatures, highlighting the potential of combining ferroelectricity and ionic conductivity for the development of advanced materials and technologies.
DOI:10.1103/PhysRevLett.131.256801
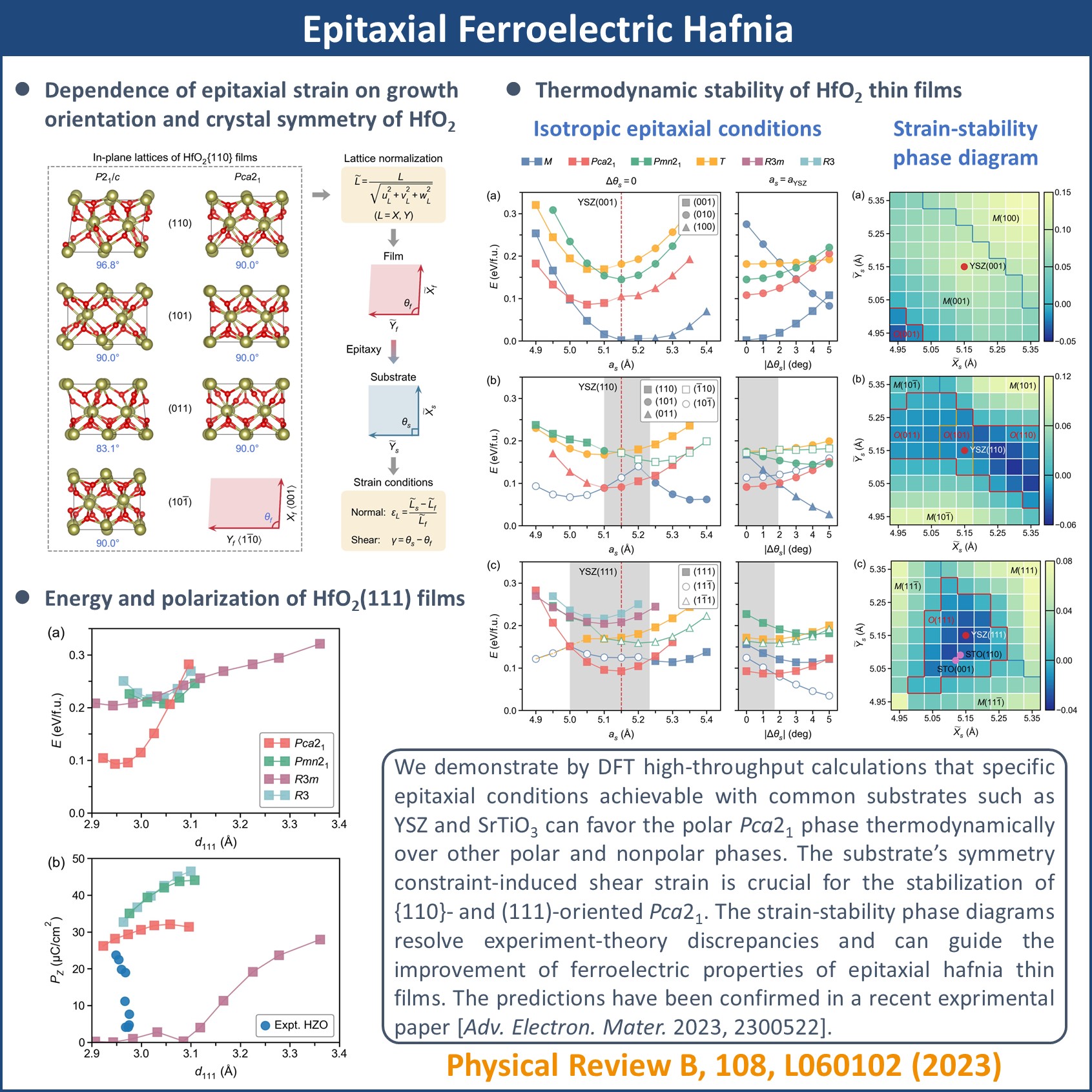
Ferroelectric memories experienced a revival in the last decade due to the discovery of ferroelectricity in HfO2-based nanometer-thick thin films. These films exhibit exceptional silicon compatibility, overcoming the scaling and integration obstacles that impeded perovskite ferroelectrics’ use in high-density integrated circuits. The exact phase responsible for ferroelectricity in hafnia films remains debated with no single factor identified that could stabilize the ferroelectric phase thermodynamically. Here, supported by density functional theory (DFT) high-throughput (HT) calculations that screen a broad range of epitaxial conditions, we demonstrate conclusively that specific epitaxial conditions achievable with common substrates such as yttria-stabilized zirconia (YSZ) and SrTiO3 can favor the polar Pca21 phase thermodynamically over other polar phases such as R3m and Pmn21 and nonpolar P21/c phase. The substrate’s symmetry constraint-induced shear strain is crucial for the preference of Pca21. The strain-stability phase diagrams resolve experiment-theory discrepancies and can guide the improvement of ferroelectric properties of epitaxial hafnia thin films.
DOI:10.1103/PhysRevB.108.L060102
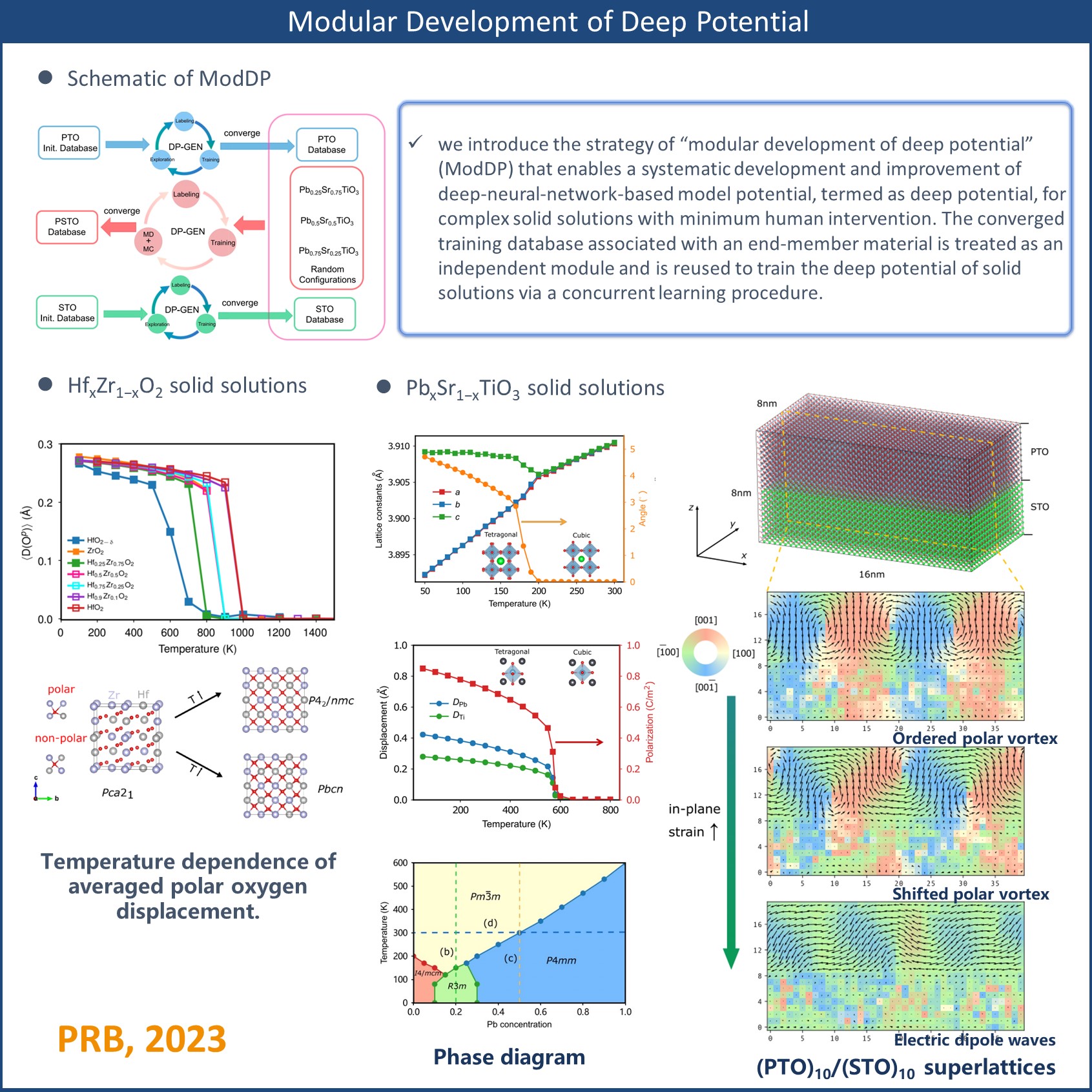
The multicomponent oxide solid solution is a versatile platform to tune the delicate balance between competing spin, charge, orbital, and lattice degrees of freedom for materials design and discovery. The development of compositionally complex oxides with superior functional properties has been largely empirical and serendipitous, in part due to the exceedingly complex chemistry and structure of solid solutions that span a range of length scales. The usage of classical molecular dynamics (MD), a powerful statistical method, in computer-aided materials design has not yet reached the same level of sophistication as that in computer-aided drug design because of the limited availability and accuracy of classical force fields for solids. Here, we introduce the strategy of “modular development of deep potential” (ModDP) that enables a systematic development and improvement of deep-neural-network-based model potential, termed as deep potential, for complex solid solutions with minimum human intervention. The converged training database associated with an end-member material is treated as an independent module and is reused to train the deep potential of solid solutions via a concurrent learning procedure. We apply ModDP to obtain classical force fields of two technologically important solid solutions, PbxSr1−xTiO3 and HfxZr1−xO2. For both materials’ systems, a single model potential is capable of predicting various properties of solid solutions including temperature-driven and composition-driven phase transitions over a wide range of compositions. In particular, the deep potential of PbxSr1−xTiO3 reproduces a few known topological textures such as polar vortex lattice and electric dipole waves in PbTiO3/SrTiO3 superlattices, paving the way for MD investigations on the dynamics of topological structures in response to external stimuli. MD simulations of HfxZr1−xO2 reveal a substantial impact of composition variation on both the phase transition temperature and the nature of the high-temperature nonpolar phase.
DOI:10.1103/PhysRevB.107.144102

Principal Investigator
Tenured Associate Professor




Address: Dunyu Road No.600, Sandun Town, Xihu District, Hangzhou, Zhejiang, China.
Copyright © 2020 developed by Yiihao HU
